Image copyright – Nature Cell Biology.
A chromatin integration labelling method enables epigenomic profiling with lower input
Published in Nature Cell Biology.
Authors: Akihito Harada, Kazumitsu Maehara, Tetsuya Handa, Yasuhiro Arimura, Jumpei Nogami, Yoko Hayashi-Takanaka, Katsuhiko Shirahige, Hitoshi Kurumizaka, Hiroshi Kimura, Yasuyuki Ohkawa
Paper presented by Dr. Atul Daiwile and selected by the NIDA TDI Paper of the Month Committee
Publication Brief Description
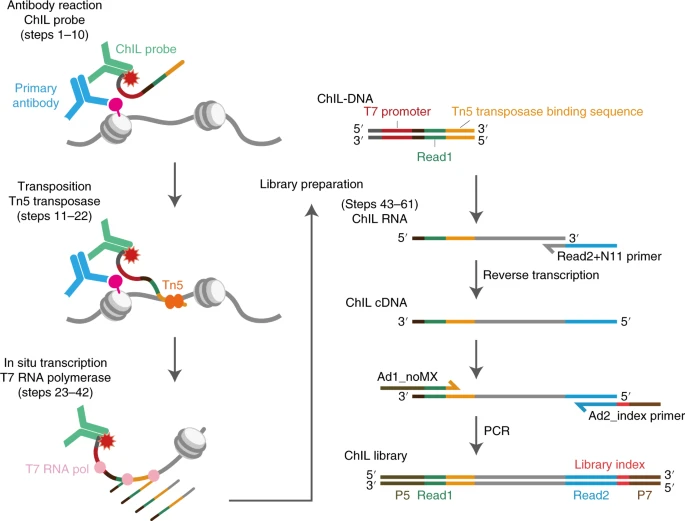
For identifying protein–DNA interactions across the genome, chromatin immunoprecipitation followed by sequencing (ChIP–seq) has been used as the standard technique. The original ChIP–seq method required typically 1 million cells. However, it is difficult to obtain epigenomic information from limited numbers of cells by ChIP–seq because of sample loss during chromatin preparation and inefficient immunoprecipitation. The ultimate goal for ChIP-Seq is to uses low-input approaches to establish single-cell epigenome/protein-binding profiling, even the best methods to date still suffer from low genome coverage. Here in the article, authors introduce chromatin integration labelling (ChIL), a technique that combines immunostaining, transposase tagging and linear amplification for low-input epigenome/protein-binding profiling even from single cells. Using ChIL followed by sequencing (ChIL–seq), authors reliably detected the distributions of histone modifications and DNA-binding factors in 100–1,000 cells. In addition, ChIL–seq successfully detected genomic regions associated with histone marks at the single-cell level. Thus, ChIL–seq can offers an alternative method to ChIP–seq for epigenomic profiling using small numbers of cell.
A chromatin integration labelling method enables epigenomic profiling with lower input Journal Article
In: Nat Cell Biol, vol. 21, no. 2, pp. 287–296, 2019, ISSN: 1476-4679.
